OpenDIA Tutorial
OpenDIA is a collection of Mass Spec Studio analysis packages and tools built on top of the OpenSWATH workflow to analyze data-independent acquisition (DIA) mass spectrometry data.
There are three major components to this new app.
1. Library creation
A new processing routine (DIA Library Creation) was developed in the Mass Spec Studio to generate configured sets of transitions, from multiple files of data-dependent acquisition data. This routine accepts raw LC-MS/MS data from multiple vendors and supports the conversion of the data into .mzml format for processing, using the MS-GF+ database search algorithm. A processing pane allows for full configuration of the search and includes parameters that anticipate the DIA experiment.
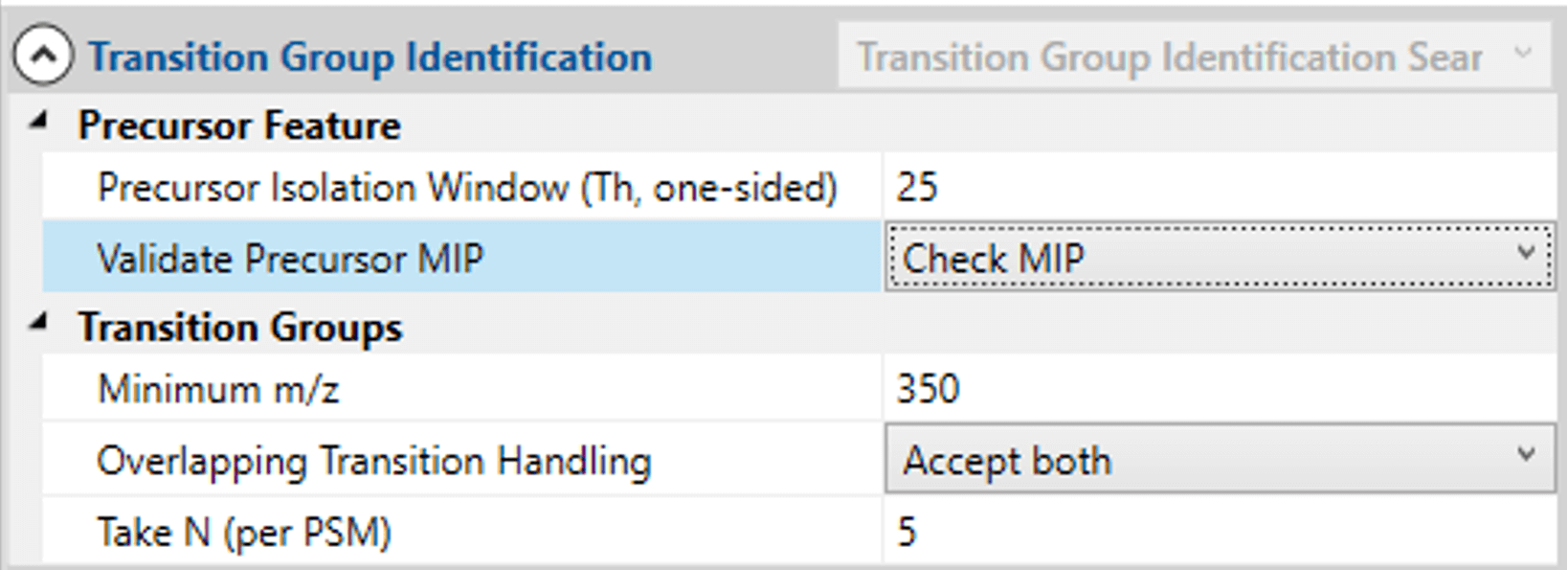
These parameters allocate peptides to DIA “bins” and specify the number of transitions that will be monitored in the DIA data analysis step.
Upon processing, the results can be inspected and navigated through interactive displays to interrogate the quality of the data used to prepare the library. All the identified peptides discovered through the search have their retention times adjusted to a unitless scale, based on the detection of reference peptides that were doped into the samples prior to analysis (iRT peptides) or discovered within the datasets themselves.
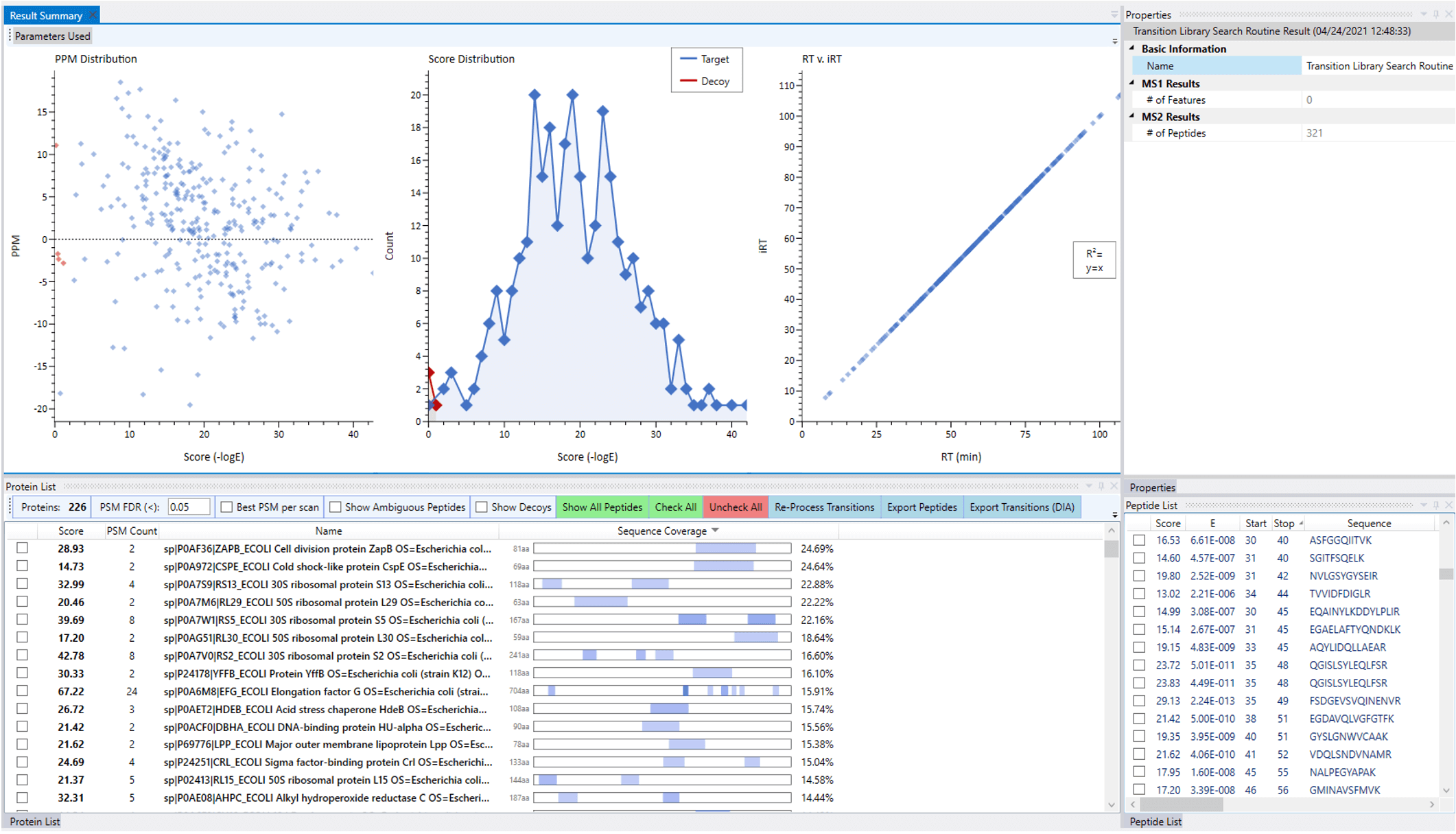
A reprocessing option allows for alternative search configurations to be considered. After optimization of the search, the resulting library is exported as a .TSV file for use in subsequent DIA experiments.
2. Library processor based on OpenSWATH
After creation of the library, DIA acquisitions can be processed using the new processing routine, called OpenDIA, built upon OpenSWATH.
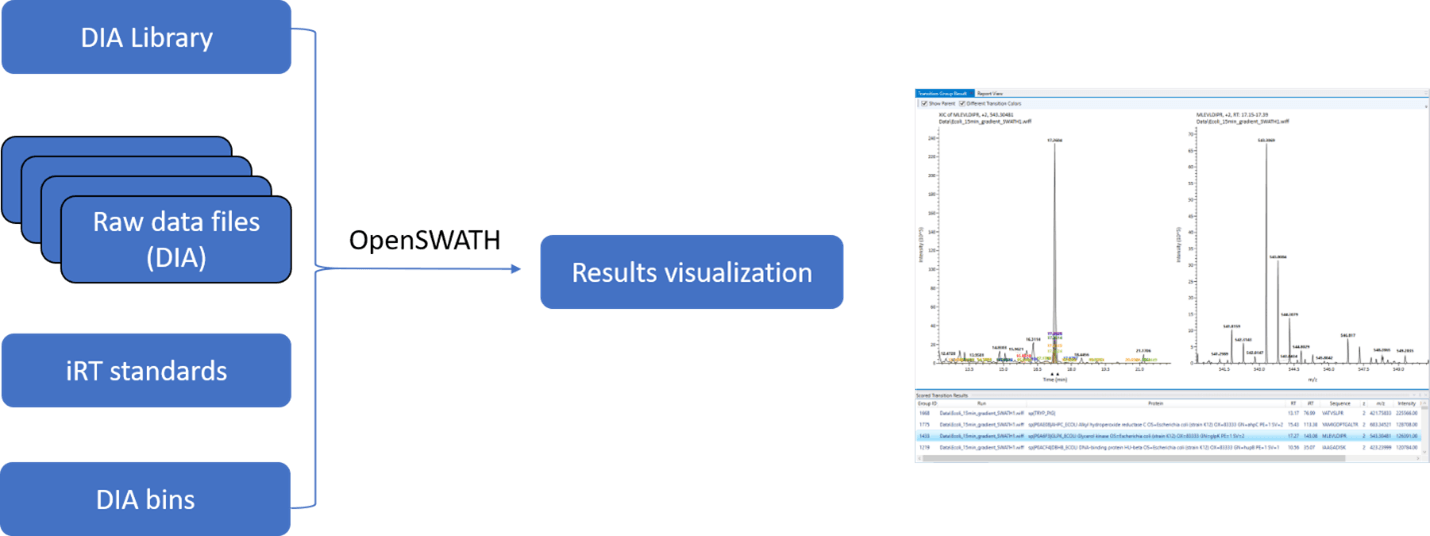
The routine is supported by a wizard that assembles all data files, along with the transitions generated in the library creation step, any peptide retention time standards that were used along with the binning of the DIA experiment itself.
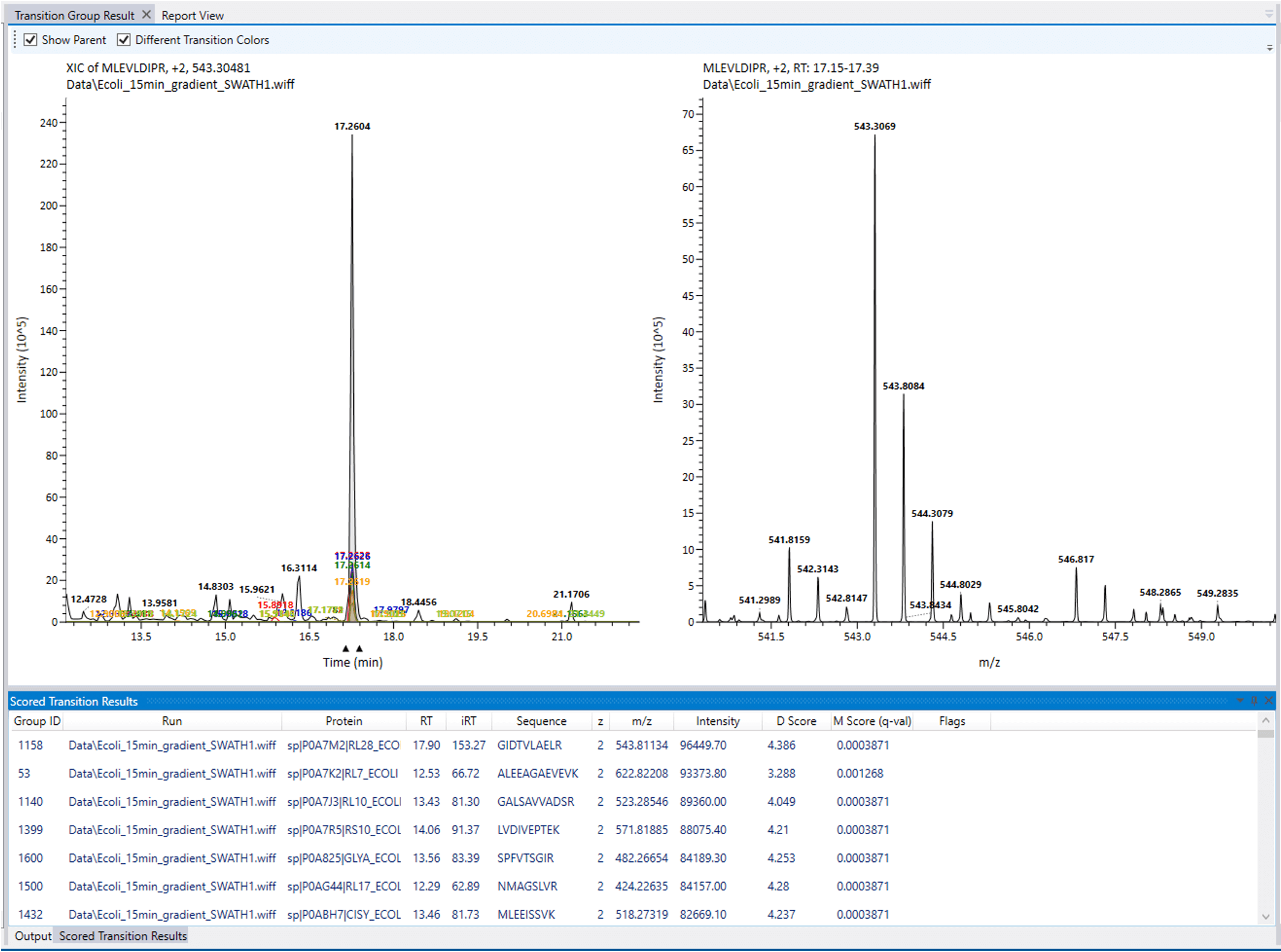
The OpenSWATH algorithms are then implemented on the files to determine scores for each transition set, based upon its semi-supervised learning and error-rate estimation routines. These scores are used to identify peptides that can support confident quantitation, assisted by an interactive data viewer. Peak intensity values are associated with all peptides.
3. Script builder for HPC
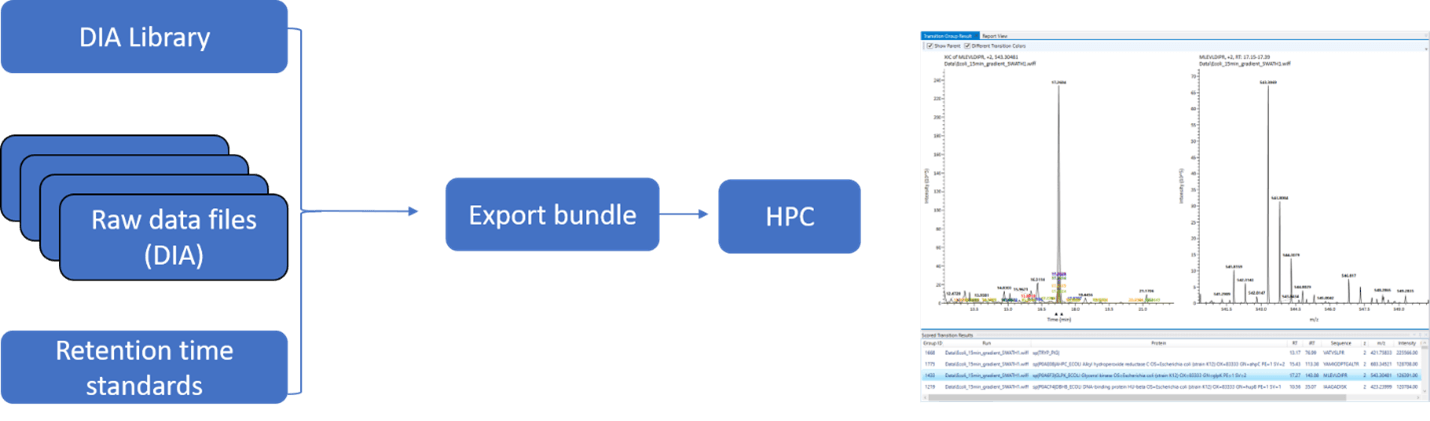
The number and size of DIA datafiles can prevent timely processing on a desktop computer. To support more efficient processing, the OpenDIA app allows for the bundling of a project that can be implemented on high-performance computing solutions, such as Compute Canada resources and Amazon Web Services.
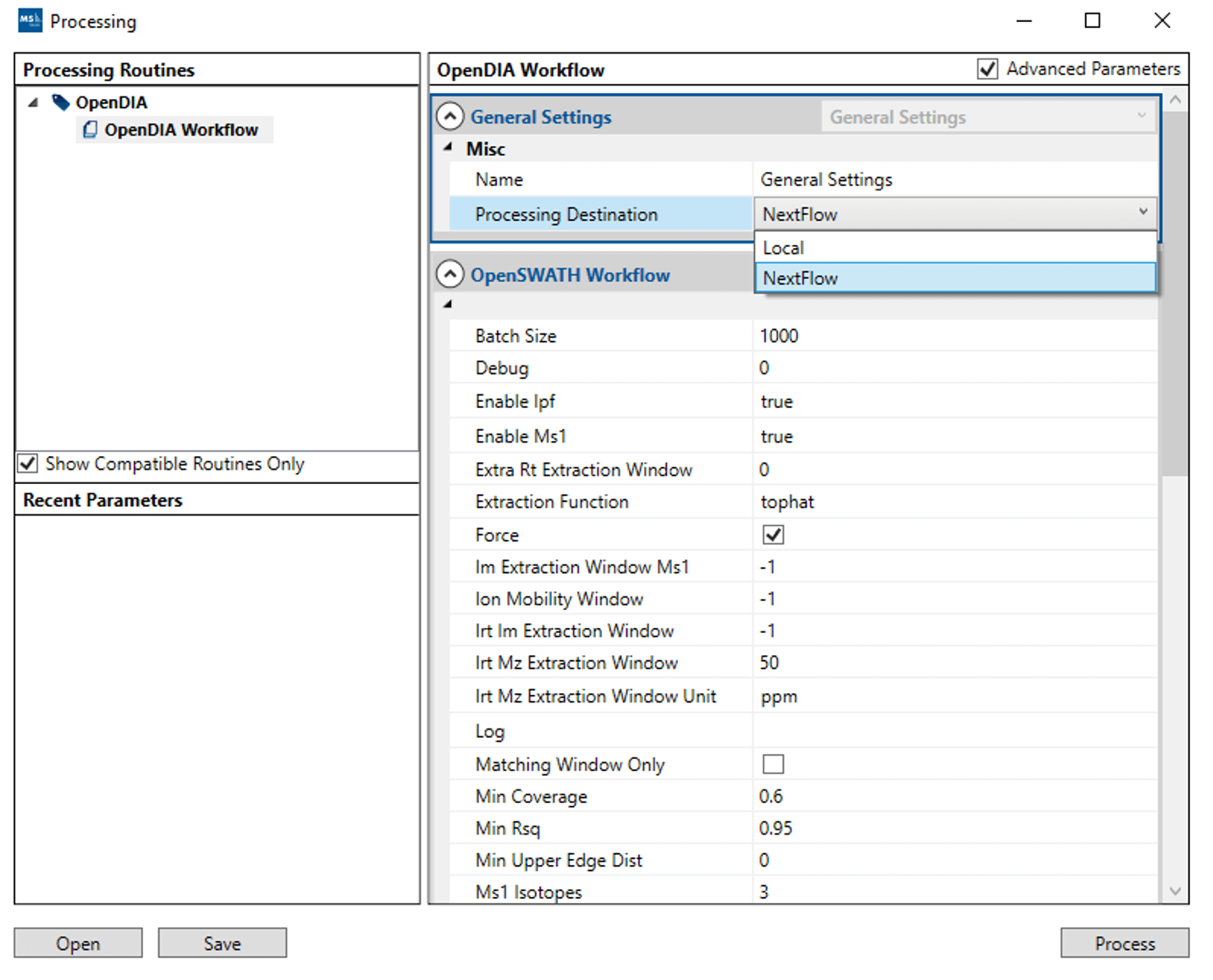
During the configuration of the processing step in OpenDIA, the user can select the routines to run on a local machine or export a bundle in a NextFlow format, to support portability and implementation on HPC schedulers and the like.
OpenPRM Tutorial
OpenPRM is a new app, paralleling OpenDIA, to promote seamless expansion of analysis to larger clinical cohorts. In many cases, a targeted analysis of select proteins identified in discovery data sets (such as in data-dependent or data-independent acquisitions) is required to simply the experiment and achieve improved sensitivity. The experiment requires identifying sets of peptides with known retention times and transitions, usually much smaller in size than the sets discovered by DIA experiments.
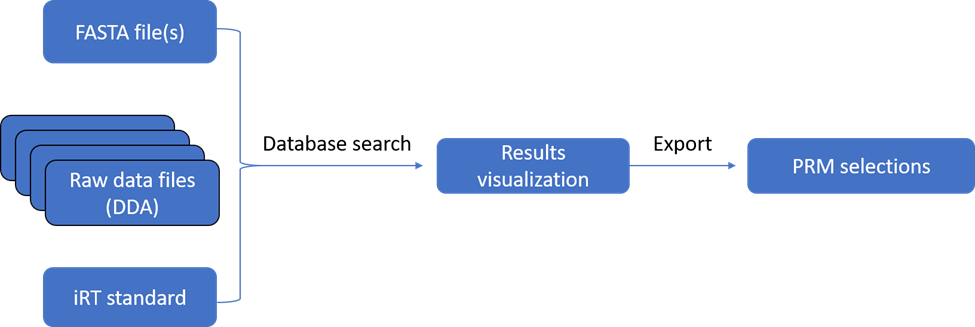
In a typical configuration, sets of data from data-dependent acquisition experiments are combined and searched in a discovery mode, using the DIA Library Creation app. The app provides the option for the inspection and selection of individual proteins and their component peptides, to aid in identifying sets of peptides for tracking in subsequent PRM experiments.
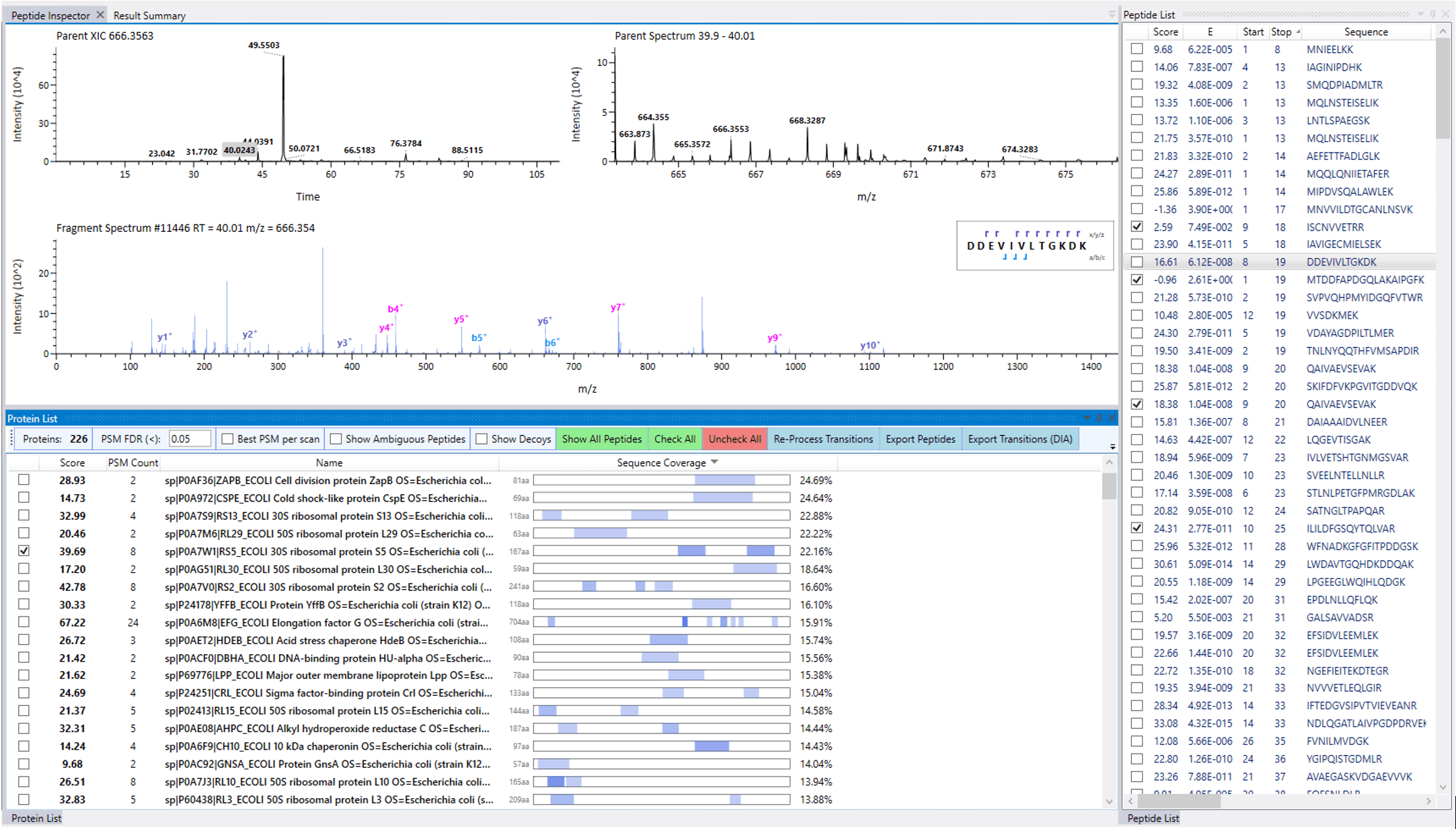
After selections are made, the user can export the peptides in a .csv format for uploading to the operating system of the chosen mass spectrometer. The normalization of the retention time scale ensures that the resulting files can be used for any LC gradient.