Hydrogen-Deuterium Exchange Analysis (PIPE/DEAL)
Mass Spec Studio v2 introduces a complete solution for HX data analysis including high-quality peptide identification, analysis, manual curation and visualization. Since v1, we introduced several exportable visualization tools, support for ion-mobility, new manual validation utilities and a brand new workflow for high-quality peptide identification.
Peptide Identification
The new Peptide Identification and Peptide Evaluation (PIPE) workflow is a recommended alternative to the proteomic-style protein identification workflows (Mascot, PEAKS, etc.). PIPE identifies the most suitable peptides for downstream HX analysis on non-deuterated data by grouping and filtering MS2 identifications based on their respective MS1 features.
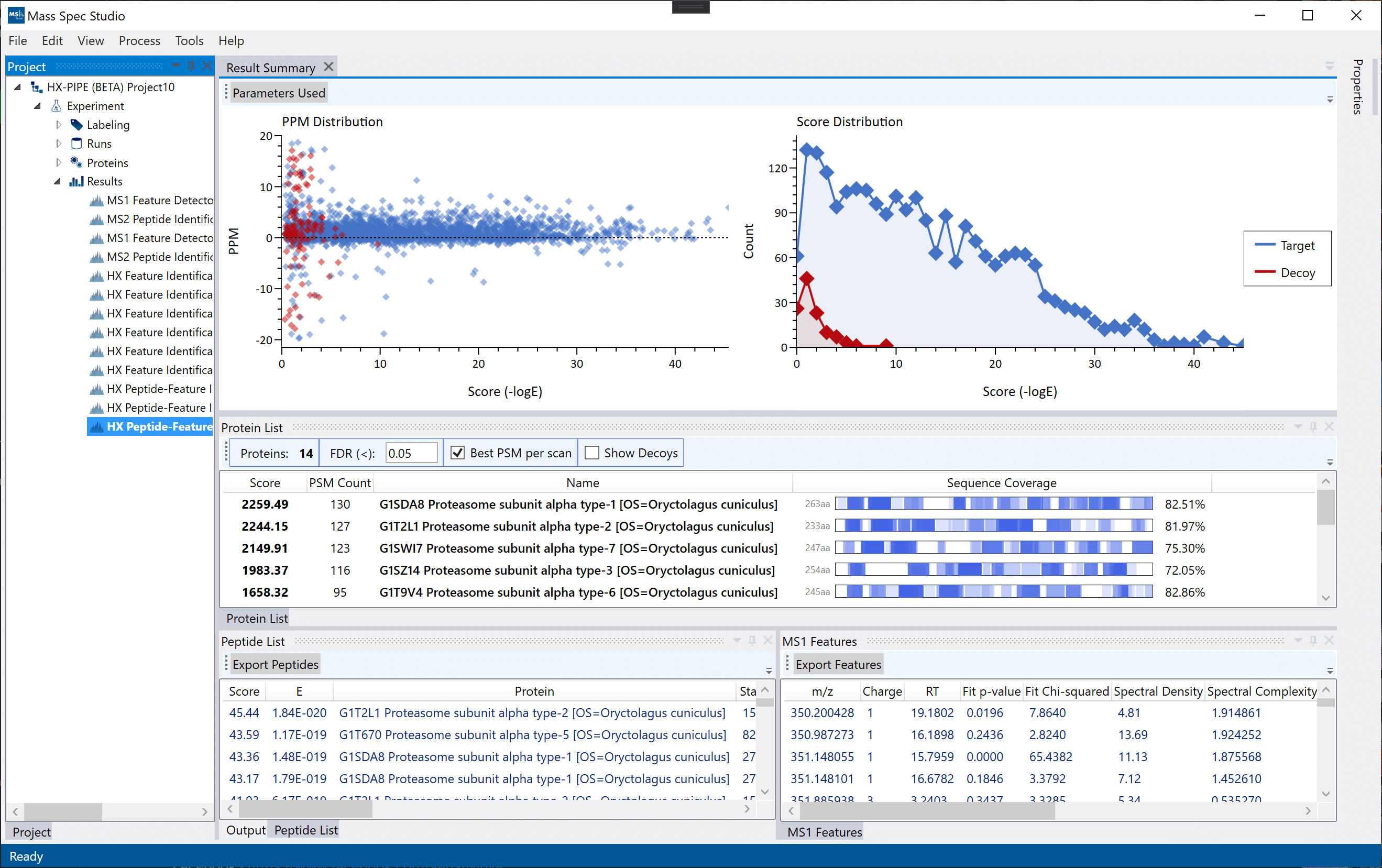
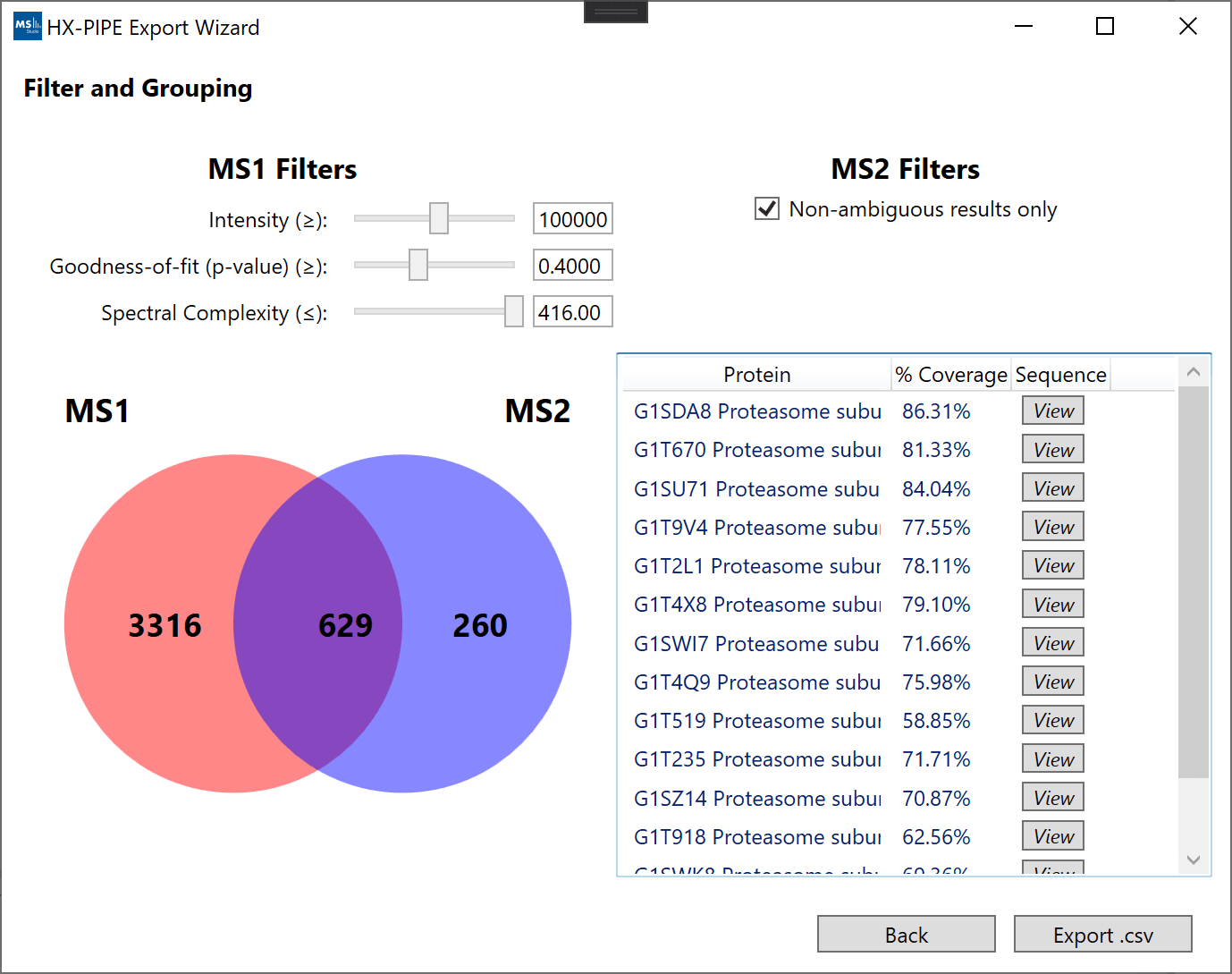
The final peptide identifications can be filtered based on the quality of their respective MS1 data using 3 filters: intensity, goodness-of-fit and spectral complexity. The peptides which pass the filters exported to be used in downstream analysis in DEAL.
Data Analysis
The Deuterium Exchange AnaLysis (DEAL) workflow has been updated with new tools and interactive visualizations supporting simple single-state HX experiments to multi-state, multi-timepoint comparative analysis. The use of PIPE for generating the peptide list is not required to use DEAL, one can still import peptide lists generated using any peptide-identification tools by converting to our input .csv format.
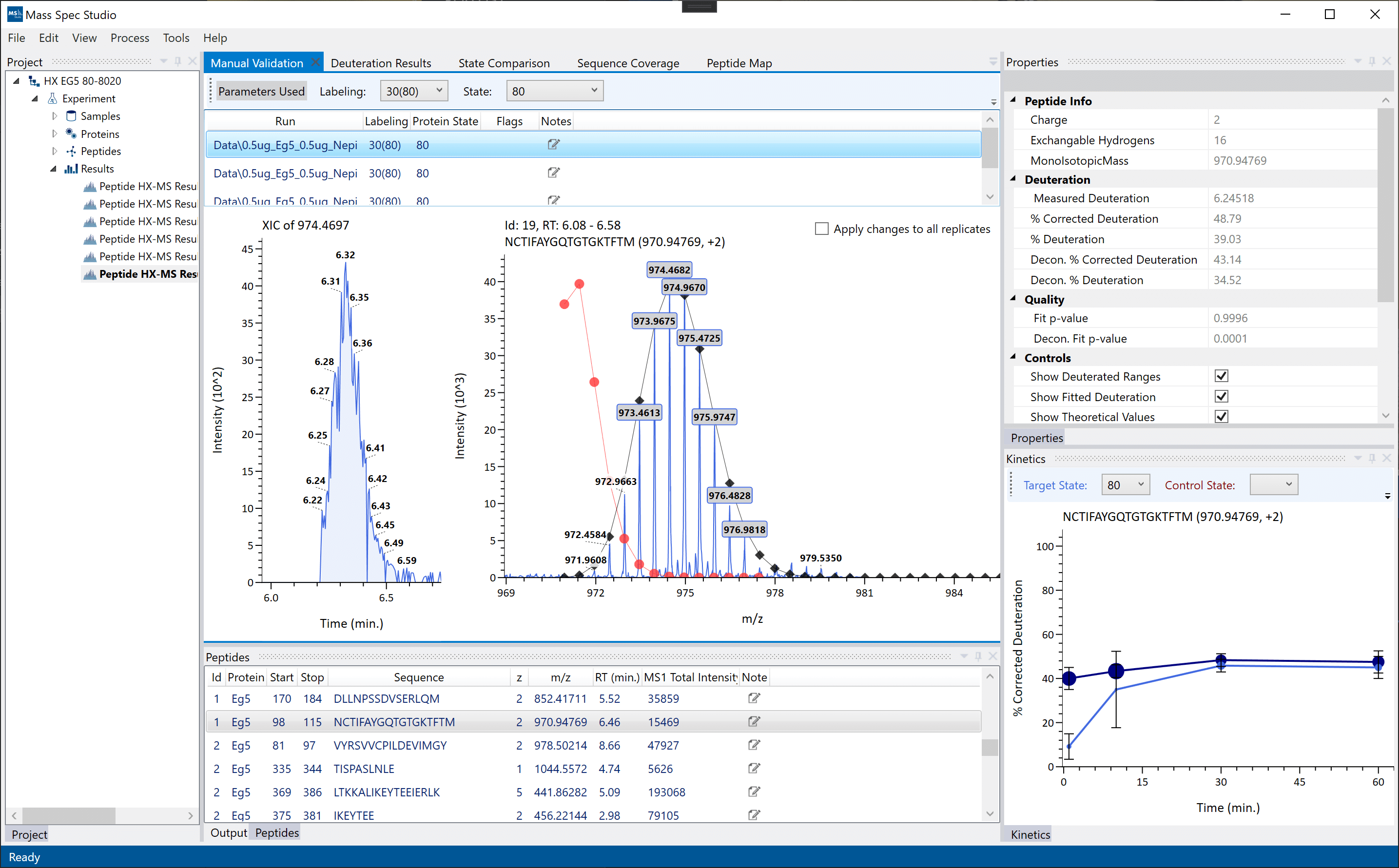
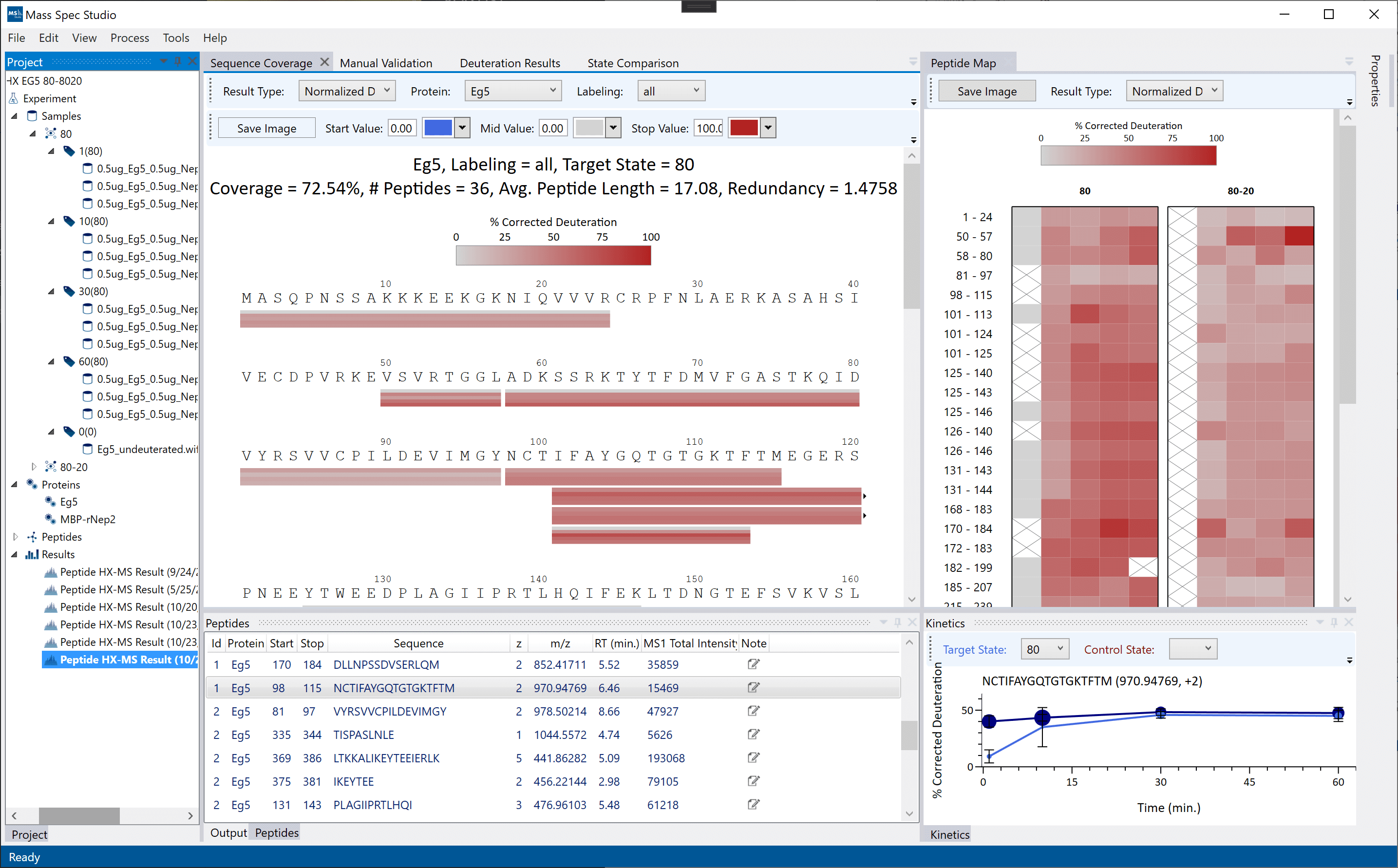
Reference
[Link, Data] Raval S, Sarpe V, Hepburn M, Crowder DA, Zhang T, Viner R, Schriemer DC. Improving Spectral Validation Rates in Hydrogen-Deuterium Exchange Data Analysis. Anal Chem, 93(9):4246-4254(2021)